Providing ab initio simulation techniques to describe the dynamics and reactions at electrified interfaces
Developing and providing accurate simulation techniques to explore and predict structural properties and chemical reactions at electrified surfaces and interfaces is critical to surmount materials-related challenges in the context of sustainability, energy conversion and storage. The groups of C. Freysoldt, M. Todorova and S. Wippermann develop various methods to incorporate finite electric fields in density-functional theory (DFT) and apply them to answer fundamental questions in corrosion, field evaporation, and the thermodynamics and transformation of electrochemical interfaces.
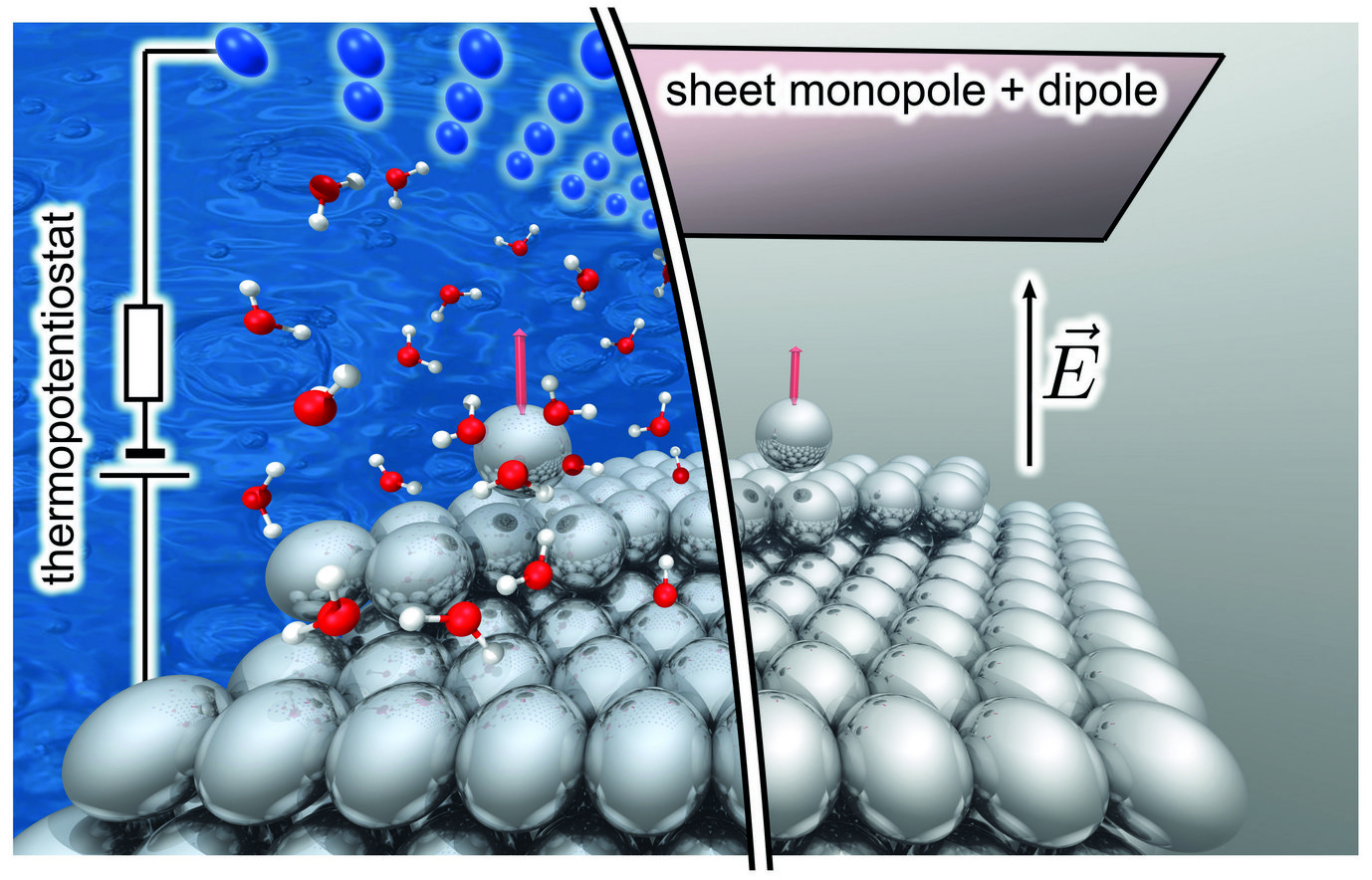
Exciting progress at the MPIE achieved over the last reporting period allows us now to realistically describe electric fields at charged surfaces from first principles. These approaches rely either on special electrostatic boundary conditions, as applied in, e.g., the modern theory of polarization, or model the electrified surface by a charged slab in a periodic cell and explicit compensating counter charges. Additionally, we introduced a “generalized dipole correction” that places an implicit computational counter electrode inside the vacuum region of the simulation cell, together with a discontinuity of the electrostatic potential [1]. The approach enables robust ab initio simulations of free surfaces in the presence of the extremely high (1011 V/m) electric fields that occur in atom probe tomography and similar experiments, and allowed us to explore and identify novel evaporation pathways [2].
When it comes to electrochemical experiments, the applied potential is held at a value that is constant on average at the macro-scale, but electronic and ionic charges fluctuate and transfer freely into and out of the region close to the interface, that is targeted by our simulations. Therefore, when studying elementary processes at electrified interfaces by DFT we must treat the local surface charge not as constant, but as a thermodynamic degree of freedom with temperature-dependent fluctuations. Building on our finite electric field techniques, we recently introduced a “thermopotentiostat”: a novel approach to control the electrode potential in ab initio molecular dynamics (AIMD) simulations [3]. By design, (i) it guarantees fundamental physical conservation laws such as the dissipation-fluctuation theorem, (ii) requires only quantities that are either easily accessible in DFT or are known from the specific computational setup, and (iii) it is straightforwardly implemented in any density-functional theory code. In fact, the thermopotentiostat has been implemented by us in VASP and in lammps.
Another technologically highly relevant field are semiconductor and (oxide) electrode materials as resulting e.g. from corrosion. Many of these materials show spontaneous polarization, which is not accounted for in conventional DFT approaches. Our recently developed generalized passivation method [4] ensures a correct description of the asymptotic bulk limit for pyroelectric materials, following a robust and quick convergence of total energies and opens the door towards an accurate description of the electronic surface band structure of such materials.
Our joint interdepartmental development activities benefit from mutual insights obtained in different areas of applications and scientific communities such as e.g. electrochemistry, surface science or semiconductor devices. In order to foster exchange also at an international level, for April 2020 we planned a three-day workshop at Ringberg castle on “Electrified solid/water interfaces – theory meets experiment”, in order to bring together leading scientists from the areas of electrochemistry, solvation and spectroscopy. Due to the CoViD-19 situation, this workshop has been rescheduled to May 2022.
 and field evaporation (right) from a surface under the influence of an applied electric field.")
2. Ashton, M. W.; Mishra, A.; Neugebauer, J.; Freysoldt, C.: Phys. Rev. Lett. 124 (2020) 176801.











