Mg Corrosion Under Anodic Conditions
We apply our novel potentiostat approach to study the chemical reactions that take place during initial corrosion at the water-Mg interface under anodic polarization. Based on the gained insight, we derive an atomistic model that explains the origin of the anodic hydrogen evolution.
Mg is a technologically highly attractive material for achieving lightweight engineering solutions, but presents a number technological challenges, such as a high vulnerability to wet corrosion. Particularly puzzling is the corrosion behaviour of Mg under anodic polarization (i.e. when Mg is positively charged), where next to extremely high corrosion rates, enhanced H2 evolution is observed [1, 2]. According to the fundamental corrosion concepts, H2 evolution should occur at the negatively charged cathode, e.g., via the reaction 2H+2e− →H2. The counterintuitive H2 reaction has been reported more than 150 years ago [3], but to this day the underlying atomistic mechanism remains elusive. We apply our novel potentiostat approach and perform ab initio molecular dynamics calculations to study the chemical reactions that occur when Mg is anodically polarized and brought into contact with water.
 open circuit conditions and (b) under anodic polarisation. Trajectories are shown in a projection along the normal to the Mgsurface. Blue, red, violet, green, and brown lines mark OH, H, H2, H+ (aq.), and O, respectively. The onset of the H2 evolution reaction is indicated in (b) by a black arrow. Tajectories of Mg atoms (intact water molecules) are shown as yellow (blue) colored background. The atomic geometry of the used supercell is visualized between the figures (a) and (b). Atoms are shown as coloredspheres: Mg (yellow), Ne (turquoise), oxygen (red).")
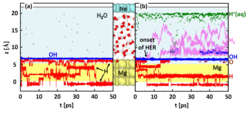
surface. Blue, red, violet, green, and brown lines mark OH, H, H2, H+ (aq.), and O, respectively. The onset of the H2 evolution reaction is indicated in (b) by a black arrow. Tajectories of Mg atoms (intact water molecules) are shown as yellow (blue) colored background. The atomic geometry of the used supercell is visualized between the figures (a) and (b). Atoms are shown as colored
spheres: Mg (yellow), Ne (turquoise), oxygen (red).
The trajectories along the interface normal are shown in Fig.1. To highlight chemical reactions trajectories of intact water molecules and of the Mg atoms are blended out. At open circuit conditions (Fig. 1(a)), i.e. without an applied electric field, we find that water molecules spontaneously dissociate at the Mg surface. The formed OH groups are adrosbed on the Mg surface and reach a maximum coverage of 1/3 monolayer. The formed H is highly mobile, does not necessarily remain on the surface, but also penetrate the electrode.
The high chemical reactivity of Mg allows us to observe under anodic polarization a surprisingly large number of reactions that take place (Fig. 1(b)), even within the short time of 50 ps we are able to access with our ab initio molecular dynamics simulation. We observe again water dissociation, but this time the OH coverage at the Mg surface reaches 1 monolayer. The H formed during the water dissociation does not all remain at Mg. We observe a field driven transfer of solvated protons (H+(aq)) towards the negatively charged Ne electrode, which occurs via a Zundel-to-Zundel Grotthus-type hopping mechanism. Under applied bias we also observe H2 evolution, which we do not see under open circuit conditions.


Inspecting the atomic geometries associated with the hydrogen evolution reaction we finde that a water molecule binds to a H atom adsorbed on the Mg surface, dissociates and leaves a H2 molecule and an OH− ion. Hereby, an unusual geometric arrangement is observed, as a H of the water molecule forms a bond with the H adsorbed on the Mg surfaces, before it dissociates from the water molecule. The only way in which the formation of such a structure can be explained is, if the H adsorbed on the Mg surface is negatively charged. Inspecting the local density of states and differences in the charge density, we find that the adsorbed H is indeed not charge neutral, but singly negatively charged H−, even under the condition of a positively charged Mg surface. The reason is the high polarizability of the Mg valence electrons, which gives rise to a large spill-out region of its electrons, causing unusual adsorption phenomena [4]. The hydrogen evolution reaction can thus be described as H−ad + H2O → H2 + OH−. In the absence of a potential (open circuit conditions), the reaction does not occur (cf. Fig. 1(a)), because the attractive electrostatic interaction between the OH− ion and the anode, which is critical to make this reaction exothermic, is absent.
Ongoing research is focusing on Mg dissolution.
References:
[1] R. Glicksman, J. Electrochem. Soc. 106, 83 (1959).
[2] N. Birbilis, A. D. King, S. Thomas, G. S. Frankel, and J. R. Scully, Electrochim. Acta 132, 277 (2014).
[3] W. Beetz, London, Edinburgh, and Dublin Philos. Mag. J. Sci. 32, 269 (1866).
[4] S.-T. Cheng, M. Todorova, C. Freysoldt, and J. Neugebauer, Phys. Rev. Lett. 113, 136102 (2014).











